













Také jste se setkali s názvy první, druhá, třetí, čtvrtá, pátá, šestá a sedmá dětská nemoc? Číslování nemocí pochází z doby, když se ještě…

Některou z očních vad trpí až 60% všech lidí. Nejčastěji se jedná o krátkozrakost, která postihuje téměř každého třetího člověka. V Evropě…
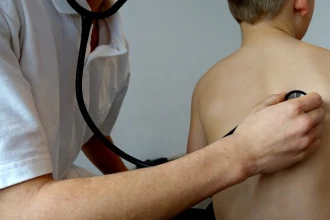
Černý kašel je vysoce infekční onemocnění, které způsobuje bakterie Bordetella pertussis. K jejímu přenosu dochází zejména kapénkovou…

Záškrt je infekční onemocnění, které způsobuje bakterie Corynebacterium diphtheriae, respektive její toxin. Tento toxin postihuje horní…
Každý týden pro vás připravujeme novou akční nabídku. Podívejte se!
Chci ušetřitHledáte všechny produkty obsahující meduňku nebo heřmánek?
Teď je najdete na jednom místě!




Abyste na našich stránkách rychle našli to, co hledáte, ušetřili spoustu klikání a nezobrazovaly se vám reklamy na věci, které vás nezajímají, potřebujeme od Vás souhlas se zpracováním souborů cookies, tj. malých souborů, které se ukládají ve vašem prohlížeči.
Podle cookies vás na našich stránkách poznáme a zobrazíme vám je tak, aby fungovaly správně a dle vašich preferencí.
Pokud nesouhlasíte, můžete
povolit pouze nezbytné
soubory cookies. Projděte si podrobný
přehled cookies a podmínky jejich užívání.
Zde máte možnost přizpůsobit soubory cookie podle kategorií, v souladu s vlastními preferencemi.
Tyto cookies jsou nezbytné kvůli správnému fungování, bezpečnosti, řádnému zobrazování na počítači nebo na mobilu, uchovávání produktů v košíku, fungujícímu vyplňování i odesílání formulářů a podobně. Technické cookies není možné vypnout, bez nich by naše stránky nefungovaly správně.
Zobrazit více Zobrazit méněČím víc lidí má statistické cookies zapnuté, tím lépe můžeme naše stránky vyladit. Třeba tak, že hojně navštěvované části stránek přesuneme hned na hlavní stránku a ušetříme tak hledání našim návštěvníkům. Díky nim jsme schopni zjistit odkud k nám lidé přicházejí, na co klikají, jak dlouho u nás zůstávají apod. Všechny informace, které soubory cookie shromažďují, jsou souhrnné a anonymní. Zpracování statistických cookies je našim oprávněným zájmem. Proti tomuto zpracování zde můžete uplatnit námitku a zastavit jejich zpracování.
Zobrazit více Zobrazit méněDíky marketingovým cookies třetích stran vám můžeme připomenout nabídky, které jste si prohlíželi na našich stránkách, i jinde na internetu: na Facebooku, na Googlu nebo třeba na Seznamu. Když tyto cookies zakážete, reklam bude pořád stejně. Ovšem na věci, které vás nezajímají. Pro dosažení tohoto účelu může docházet k profilování. Tyto cookies zpracováváme na základě vašeho souhlasu.
Zobrazit více Zobrazit méněNejdříve se musíte přihlásit.
DOPRAVA ZDARMA
k nákupu nad 599 Kč
Přihlaste se k odběru novinek
a slevový kód vám pošleme e-mailem.







